Faculty Profile
 Brad P. Carrow
Brad P. Carrow
Associate Professor
Department of Chemistry
Office: STL, 453
Contact: bcarrow@central.uh.edu - 713-743-2569
Overview
We are interested in the design of transition metal catalysts that can enable broadly applicable organic transformations and address challenges in sustainability and materials chemistry. Our approach is interdisciplinary, bridging the fields of organometallic, physical organic, synthetic organic, and polymer chemistry. A core emphasis in our group is the mechanism-informed design of ligands and metal complexes that can manifest new elementary catalytic steps, control speciation between active and inactive catalyst states, and enable new classes of substrate to be integrated into synthetic organic chemistry.
New Elementary Organometallic Mechanisms
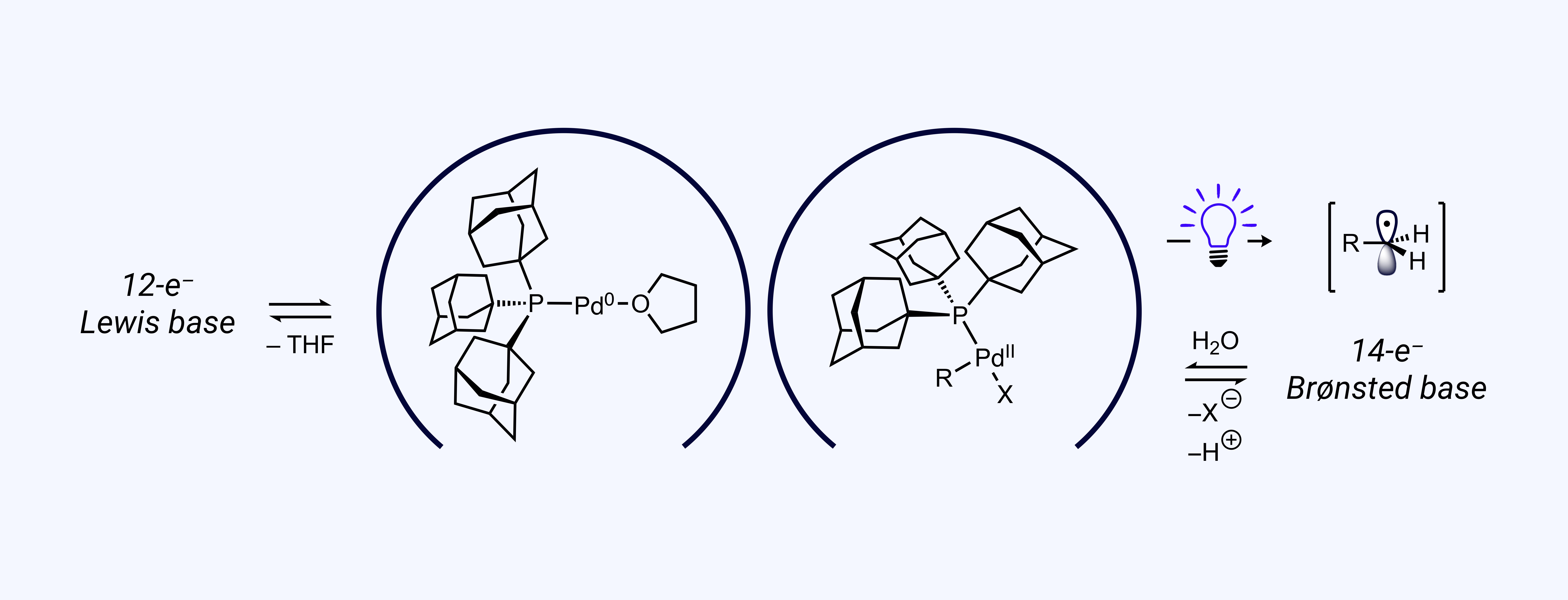
Special things can happen when a metal catalyst is driven toward a persistent, coordinatively unsaturated state. A new family of ligands we have developed, the diamondoid phosphines, are particularly well suited for this task because of their size, chemical stability, and large polarizability. Recently, we have found complexes with a diamondoid phosphine can undergo “soft deprotonation” of latent nucleophiles, such as water, to generate nucleophilic intermediates under neutral to acidic conditions. This mechanism can enable transmetalation in C–C and C–X cross-coupling reactions under exceptionally mild conditions compatible with the most sensitive substrates. We have also observed visible light-promoted bond weakening in related complexes, which enables single electron catalytic processes under ambient conditions with metals not able to do so classically. Low oxidation state, low coordinate metal species can also be generated with implications for studying and tuning site selective catalysis involving oxidative addition.
Design Concepts for Catalysts in Central Synthetic Transformations
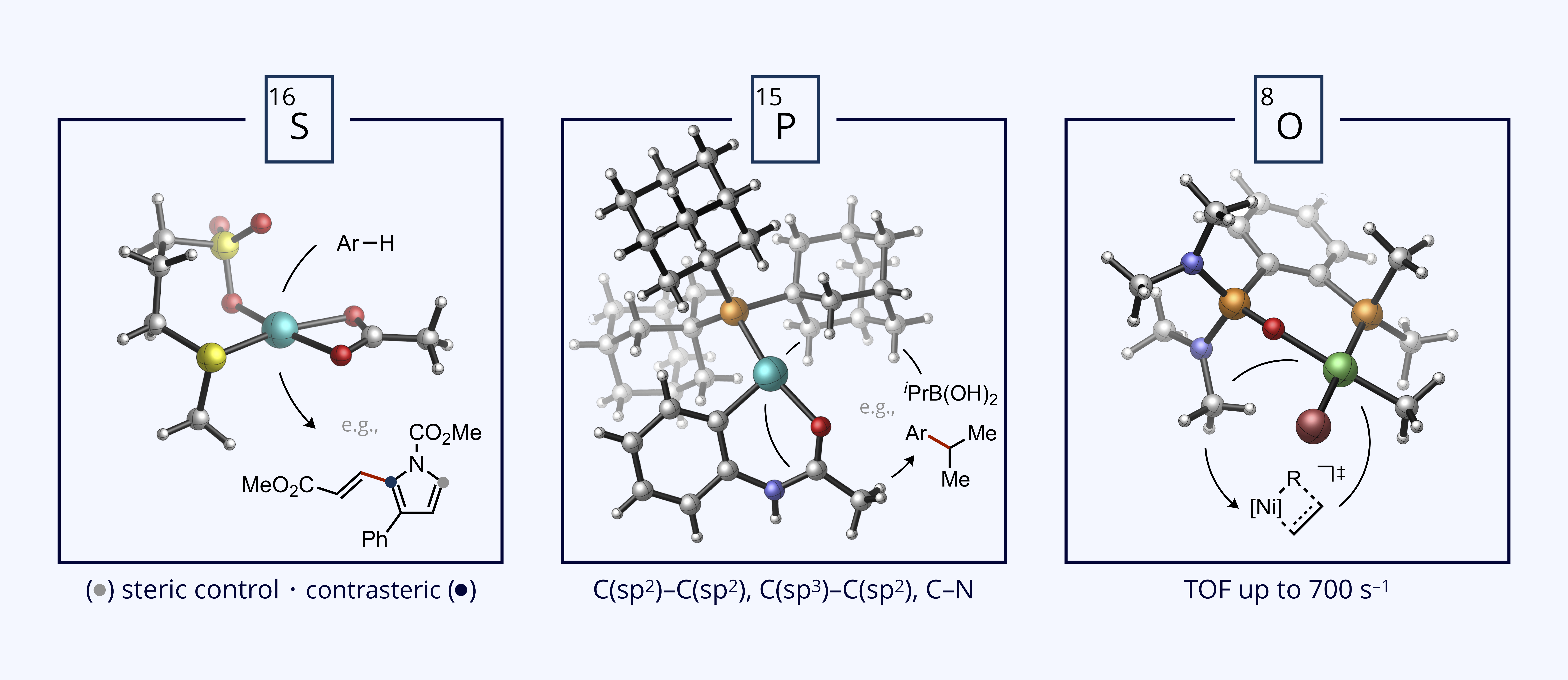
The need for an oxidant to thermodynamically drive dehydrogenative coupling demands access to new ligand classes beyond those developed for inert conditions (i.e., metathesis, cross-coupling, hydrogenation). We have developed a class of simple thioethers that can substantially accelerate cross-dehydrogenative coupling, which is correlated to a more asynchronous but concerted mechanism for C–H activation termed eCMD. The ubiquity of thioethers in commercial and natural building blocks represents a broad and underutilized chemical space for tuning of chemo-, site-, and stereoselectivity during oxidative C–H functionalization reactions.
We are also interested in incorporating polar functionality into polyolefins in a practical fashion, which remains a long-standing unsolved problem in polymer synthesis. A seeming paradox to this end: functional group tolerance is improved with decreasing catalyst electrophilicity, but insertion polymerization strongly favors more electrophilic metals. We have developed a modular ligand class based on a P(V) oxide donor with considerable through-bond electron donation (lower Lewis acidity) but also manifests increased partial positive charge on the metal through electrostatic effects to stabilize the polarized migratory insertion transition state to allow for fast propagation.
Merging Classically Incompatible Building Blocks for New Materials
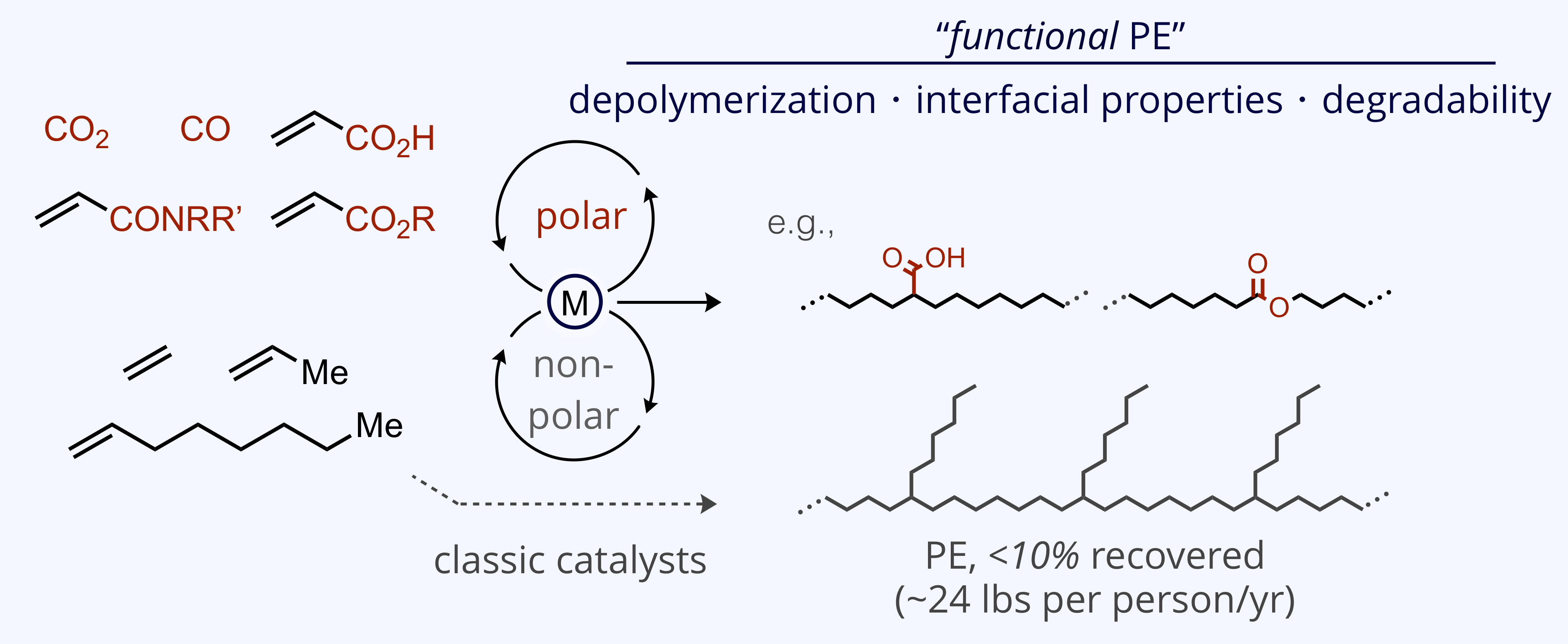
The plastics economy of the future will need to address issues of pollution, sustainability, and advanced material properties not currently accessible with 20th century thermoplastic materials. We are pursuing catalytic strategies to enchain historically incompatible mixtures of non-polar with polar C1 & C2 monomers to create functional polymers with enhanced properties that can help to address these issues.
- Assistant Professor, University of Tokyo, 2011-2013
- Postdoctoral Fellow with Kyoko Nozaki, University of Tokyo, 2011
- Ph.D. with John F. Hartwig, University of Illinois at Urbana-Champaign, 2011
- B.S. in Chemistry, Missouri University of Science and Technology, 2003